Unlocking Clean and Safe Energy for Future Vehicles
Other Articles

Advanced Cobalt-Based Catalysts Cut Costs and Supercharge Efficiency in Hydrogen Fuel Cell Vehicles
Study conducted by Prof. Molly Mengjung LI  and her research team
and her research team

The global transition towards sustainable energy has placed hydrogen-powered vehicles at the forefront of clean transportation solutions. As governments and industries strive to decarbonise mobility, the acceptance of hydrogen fuel cell vehicles is gaining momentum due to their high energy efficiency and zero-emission credentials. However, the widespread adoption of hydrogen energy vehicles hinges not only on the development of fuel cell technology but also on the safe, efficient, and cost-effective storage and release of hydrogen itself.
Prof. Molly Mengjung LI, Assistant Professor of the Department of Applied Physics at The Hong Kong Polytechnic University, and her research team are investigating the possibility of using ammonia as a hydrogen fuel carrier and studying the stability of hydrogen energy storage in order to promote the popularisation of hydrogen-powered vehicles. Their study, published in Advanced Materials, introduces an efficient and cheap catalyst to facilitate the hydrogen energy generation reaction [1].
Hydrogen (H2), when used in fuel cells, reacts with oxygen (O2) to generate electricity, emitting only water (H2O) as a by-product. This reaction offers a compelling alternative to fossil fuel combustion, promising both environmental and operational advantages. However, hydrogen’s low volumetric density and the challenges associated with its storage and transport have long been recognised as significant barriers to its practical deployment. Among the various strategies proposed, chemical carriers such as ammonia (NH3) have emerged as promising solutions. NH3 boasts a well-established production and distribution infrastructure, a high hydrogen density and the ability to release hydrogen without generating carbon oxides. The decomposition of NH3 into N2 and H2 is thus a critical reaction for on-board hydrogen generation in fuel cell vehicles.
Despite its promise, the practical implementation of NH3 cracking technology faces a major hurdle—the reliance on ruthenium (Ru)-based catalysts. Ru catalysts are highly effective for low-temperature NH3 decomposition but their scarcity and high cost impede large-scale adoption. This has spurred a global research effort to identify alternative catalysts based on earth-abundant, non-noble metals. Cobalt (Co) has emerged as a particularly attractive candidate, given its favourable nitrogen binding energy and lower susceptibility to catalyst poisoning compared to other transition metals. However, conventional Co-based catalysts typically require high temperatures (>600°C) to achieve satisfactory hydrogen yields, limiting their utility for mobile applications where energy efficiency and compact reactor design are paramount considerations.
To address these challenges, recent research has focused on innovative catalyst design strategies that can enhance the low-temperature activity of Co-based systems. One such approach is the engineering of lattice strain at the catalyst-support interface, which can modulate the electronic structure of active sites and thereby optimise their interaction with reactants. Drawing inspiration from advances in strain engineering in other catalytic systems, Prof. Li’s research team has developed a new class of core@shell catalysts, exemplified by the Co@BaAl₂O₄₋ₓ heterostructureNote1 (Figure 1a).
The rationale for selecting the Co@BaAl₂O₄₋ₓ architecture lies in its ability to harness strong metal-support interactions (SMSI) and induce tensile strain at the interface between the cobalt core and the barium aluminate shell. This strained heterostructure is synthesised via a Tammann temperature-guided co-precipitation method, followed by controlled calcination and reduction steps. The resulting catalyst features cobalt nanoparticles (average size ~14 nm) (Figure 1b) encapsulated within a thin (3 nm) BaAl₂O₄₋ₓ shell (Figure 1c), which is rich in oxygen vacancies. These vacancies not only facilitate reactant permeability but also play a crucial role in the catalytic process.
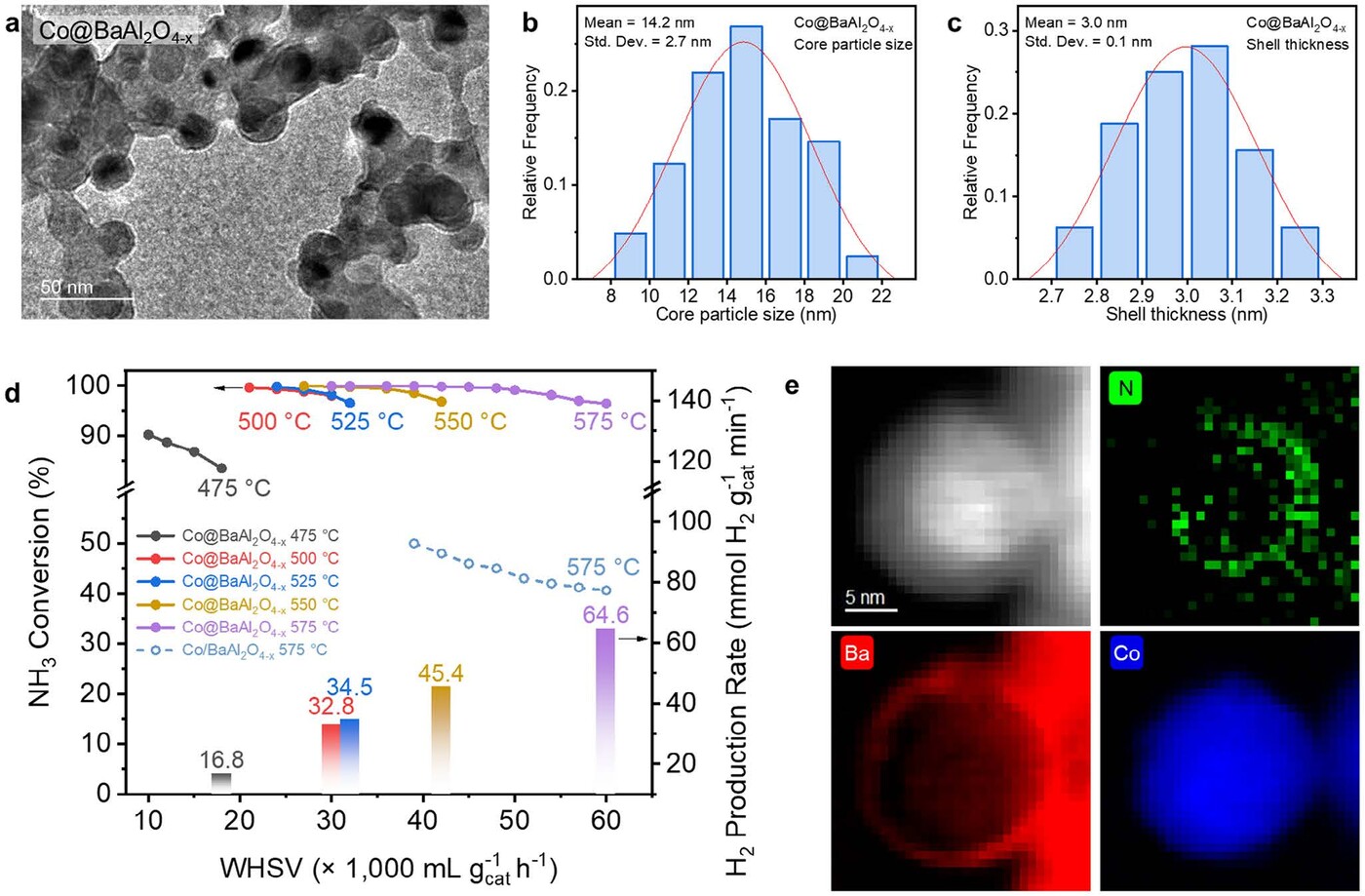
Figure 1. SMSI core@shell catalyst composition optimisation for NH3 decomposition reaction
a) Co@BaAL2O4-x heterostructure under transmission electron microscope
b-c) Core@shell heterostructure with an average Co particle size of 14.2±2.7 nm and a shell thickness of 3.0±0.1 nm
d) NH3 conversion and corresponding H2 production rate as a function of WHSV over Co@BaAL2O4-x and Co/BaAL2O4-x
e) Scanning transmission electron microscopy images and electron energy loss spectroscopy element distribution maps of post-reaction Co/BaAL2O4-x, showing the accumulation of nitrogen species at the core@shell interface
Performance testing of the Co@BaAl₂O₄₋ₓ catalyst reveals remarkable activity for NH3 decomposition at moderate temperatures. Under high space velocity conditions, the catalyst achieves a hydrogen production rate of 64.6 mmol H₂ gcat-1 min-1 and maintains nearly complete NH3 conversion between 475°C and 575°C (Figure 1d). These results are on par with, or even surpass, those of many Ru-based catalysts, but without the associated cost and supply constraints. Advanced characterisation techniques, including synchrotron X-ray absorption spectroscopy and electron microscopy, confirm the formation of a well-defined core@shell structure and the presence of nitrogen species at the interface after reaction (Figure 1e), highlighting the critical role of the heterostructure in facilitating the catalytic process.
To further elucidate the advantages of the core@shell design, a comparative study was conducted with a conventional supported catalyst, Co/BaAl₂O₄₋ₓ, which lacks the encapsulating shell. Both catalysts were prepared with similar cobalt nanoparticle sizes to ensure a fair comparison. The results are striking: while both systems exhibit increasing NH3 conversion with temperature, the core@shell Co@BaAl₂O₄₋ₓ catalyst demonstrates a significantly lower onset temperature for activity (200°C versus 250°C) and achieves near-complete conversion at 500°C, compared to 550°C for the supported analogue (Figure 1d). Moreover, the core@shell structure exhibits superior stability under high flow rates, whereas the supported catalyst suffers from a sharp decline in performance.
Kinetic analyses provide further insights into the origins of this enhanced activity. The apparent activation energy for NH3 decomposition over Co@BaAl₂O₄₋ₓ is measured at 64.8 kJ mol-1, closely matching the best Ru-based systems, while the supported catalyst exhibits a much higher barrier (82.3 kJ mol-1). Reaction order studies indicate that the core@shell configuration promotes stronger NH3 adsorption and more efficient H2 removal, mitigating the limitations typically encountered with conventional Co catalysts. Temperature-programmed surface reaction and in situ infrared spectroscopy corroborate these findings, revealing enhanced NH3 adsorption and facilitated N–H bond cleavage at the heterostructure interface.
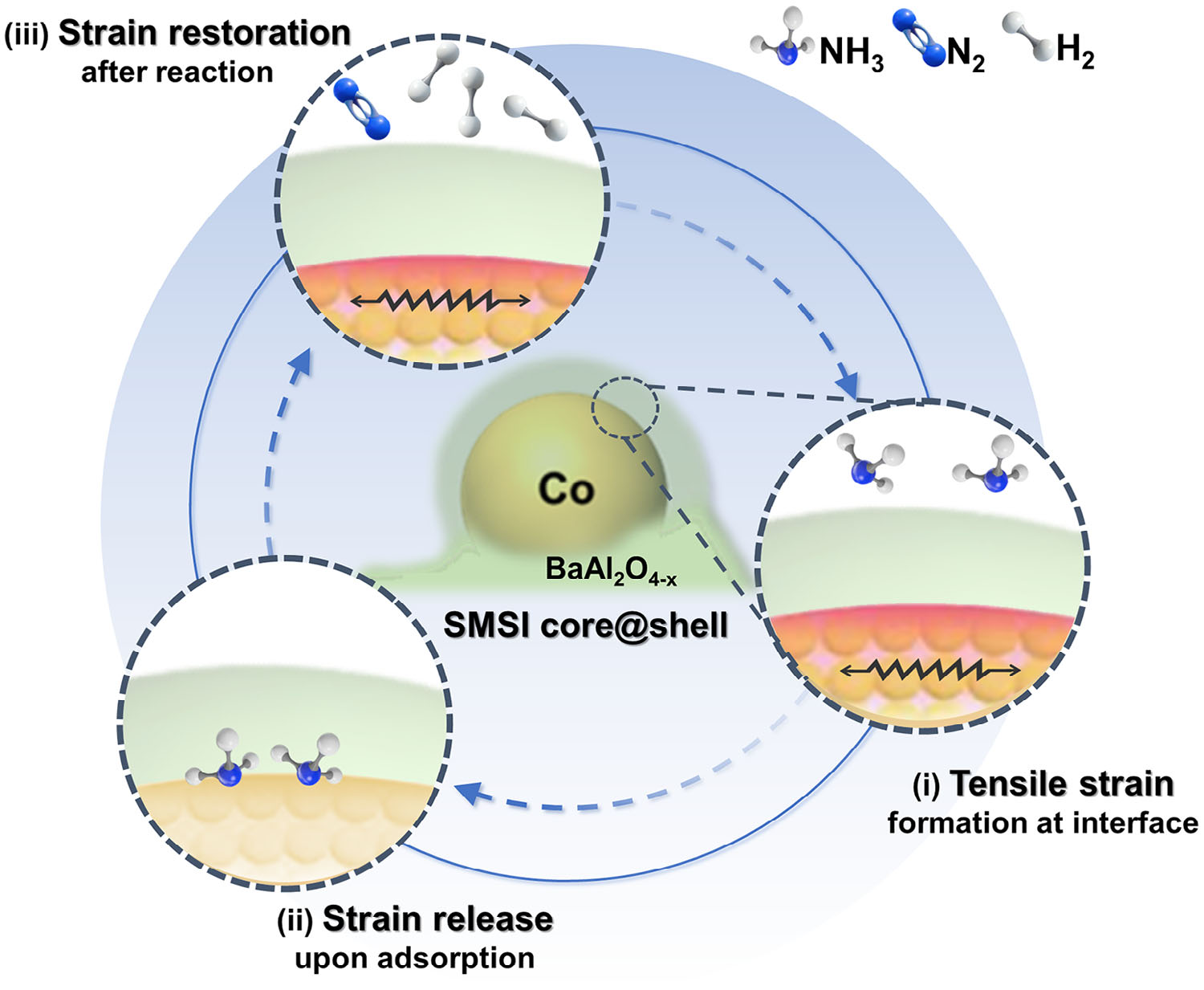
Figure 2. The dynamic lattice strain evolution mechanism for HN3 decomposition over Co@BaAl2O4-x catalyst
The key to the superior performance of the Co@BaAl₂O₄₋ₓ catalyst lies in the dynamic evolution of lattice strain at the core@shell interface (Figure 2). Synchrotron X-ray diffraction and electron microscopy reveal that the cobalt lattice is expanded at the interface, corresponding to a tensile strain of approximately +3.2%. This strain modulates the electronic structure of cobalt, as evidenced by X-ray absorption and photoelectron spectroscopy, which show a higher valence state and a positive shift in the d-band centre. Density functional theory calculations confirm that the strained Co surface exhibits an upward shift in the d-band centre, enhancing its affinity for NH3 and facilitating the rate-determining N–H bond dissociation step.
Crucially, this lattice strain is not static but evolves dynamically during the catalytic cycle. Upon NH3 adsorption, the tensile strain is partially released, leading to a downward shift in the d-band centre and a reduction in adsorption strength. This facilitates the desorption of reaction products (H2 and N2), preventing the accumulation of intermediates that could poison active sites. Subsequent desorption restores the tensile strain, reactivating the interface for the next cycle. This dynamic strain evolution mechanism ensures sustained catalytic activity and high efficiency, even under demanding reaction conditions.
The development of the Co@BaAl2O4-x core@shell catalyst represents a significant advance in the quest for efficient, Ru-free catalysts for ammonia cracking in hydrogen energy vehicles. By leveraging lattice strain engineering and strong metal-support interactions, this system achieves low-temperature activity and stability previously attainable only with precious metals. The mechanistic insights gained from this work not only inform the design of next-generation catalysts for clean energy applications but also underscore the transformative potential of interface engineering in heterogeneous catalysis. As the hydrogen economy continues to evolve, such innovations will be pivotal in realising the full potential of hydrogen as a sustainable fuel for the future of mobility.
Prof. Li is currently an Assistant Professor of the Department of Applied Physics at The Hong Kong Polytechnic University. She has a solid foundation and wide experience in the chemistry of materials and applied catalysis in green energies and the environment, specifically focusing on the design of efficient catalyst systems for turning waste to renewable fuels as well as the reversible hydrogen storage cycle based on methanol or ammonia conversion.
| Notes |
|---|
A core@shell structure is a type of material design comprising an inner core of one substance, surrounded by an outer shell of another substance. The suggested catalyst Co@BaAl2O4-x consists of Co as the core and BaAl2O4-x as the shell. “4 – x” in BaAl2O4-x indicates the oxygen vacancies in the BaAl2O4.
| References |
|---|
[1] Xiong, P., Li, J., Xu, Z., Lin, Y., Bennett, R. D., Zhang, Y., Tu, W., Zhu, Y., Soo, Y., Wu, T., & Li, M. M. (2025). Efficient Low‐temperature Ammonia Cracking Enabled by Strained Heterostructure Interfaces on Ru‐free Catalyst. Advanced Materials. https://doi.org/10.1002/adma.202502034
 | Prof. Molly Mengjung LI |


