

Qin, Y., Wang, Y., Lu, J., Xu, L., & Wong, W.-Y. (2024). A Highly Conjugated Nickel(II)-Acetylide Framework for Efficient Photocatalytic Carbon Dioxide Reduction. Angewandte Chemie International Edition, 2025, 64, e202418269.
https://doi.org/10.1002/anie.202418269
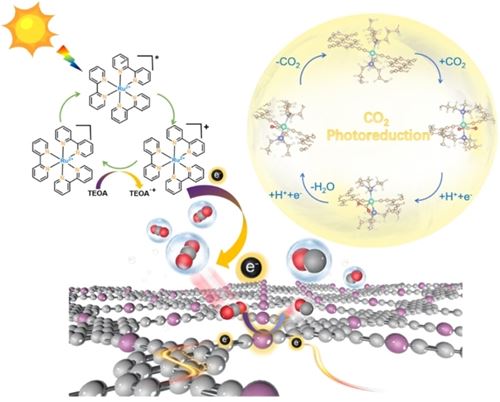
The incorporation of transition-metal single atoms as molecular functional entities into the skeleton of graphdiyne (GDY) to construct novel two-dimensional (2D) metal-acetylide frameworks, known as metalated graphynes (MGYs), is a promising strategy for developing efficient catalysts, which can combine the tunable charge transfer of GDY frameworks, the catalytic activity of metal and the precise distribution of single metallic centers. Herein, four highly conjugated MGY photocatalysts based on NiII, PdII, PtII, and HgII were synthesized for the first time using the ‘bottom-up’ strategy through the use of M−C bonds (−C≡C−M−C≡C−). Remarkably, the NiII-based graphyne (TEPY-Ni-GY) exhibited the highest CO generation rate of 18.3 mmol g−1 h−1 and a selectivity of 98.8 %. This superior performance is attributed to the synergistic effects of pyrenyl and −C≡C−Ni(PBu3)2−C≡C– moieties. The pyrenyl block functions as an intramolecular π-conjugation channel, facilitating kinetically favorable electron transfer, while the −C≡C−Ni(PBu3)2−C≡C− moiety serves as the catalytic site that enhances CO2 adsorption and activation, thereby suppressing competitive hydrogen evolution. This study provides a new perspective on MGY-based photocatalysts for developing highly active and low-cost catalysts for CO2 reduction.
Sun, M., Wong, H. H., Wu, T., Dougherty, A. W., Huang, B., Entanglement of Spatial and Energy Segmentation for C1 Pathways in CO2 Reduction on Carbon Skeleton Supported Atomic Catalysts. Adv. Energy Mater. 2022, 12, 2103781.
https://doi.org/10.1002/aenm.202103781
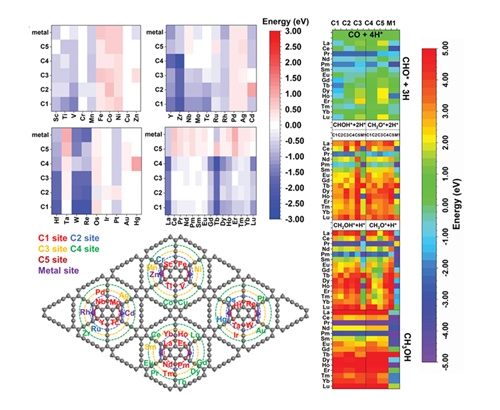
Electroreduction of CO2 has become the most attractive approach to generate value-added chemicals and fuels. Products of single atomic catalysts (SAC) in CO2 reduction reaction reactions (CO2RR) are mostly limited to CO since the contributions of spatial and thermodynamic factors are not distinguished. To break through the challenges, comprehensive explorations in graphdiyne(GDY)-based SAC are made, to reveal detailed influences of active sites, elements, and adsorptions on the selectivity and reaction energy of the C1 pathway. Unique d electrons dominated adsorption behaviors are identified, where the d6 boundary is able to help screen out promising candidates for achieving complicated C2+ products. Based on spatial and thermodynamic factors, metal sites are still the most promising active sites. The transition metal based GDY-SACs show element-dependent electroactivity towards different products in CO2RR. Meanwhile, the GDY-Pr and GDY-Pm SACs are promising candidates for the CO2RR and even C2 products in the future. This work supplies in-depth insights into the CO2RR to facilitate the design of efficient atomic catalysts in future work.
Sun, M., Dougherty, A. W., Huang, B., Li, Y., Yan, C.-H., Accelerating Atomic Catalyst Discovery by Theoretical Calculations-Machine Learning Strategy. Adv. Energy Mater. 2020, 10, 1903949.
https://doi.org/10.1002/aenm.201903949
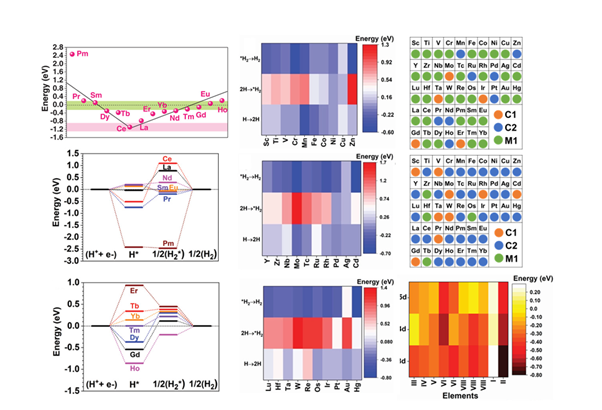
In this work, the systematic investigation of the HER process in graphdyine (GDY) based AC is presented in terms of the adsorption energies, adsorption trend, electronic structures, reaction pathway, and active sites. This comprehensive work innovatively reveals GDY based AC for HER covering all the transition metals (TM) and lanthanide (Ln) metals, enabling the screening of potential catalysts. The density functional theory (DFT) calculations carefully explore the HER performance beyond the comparison of sole H adsorption. Therefore, the screened catalysts candidates not only match with experimental results but also provide significant references for novel catalysts. Moreover, the machine learning (ML) technique bag-tree approach is innovatively utilized based on the fuzzy model for data separation and converse prediction of the HER performance, which indicates a similar result to the theoretical calculations. From two independent theoretical perspectives (DFT and ML), this work proposes pivotal guidelines for experimental catalyst design and synthesis. The proposed advanced research strategy shows great potential as a general approach in other energy-related areas.